Next: 8 Session information Up: MANOR: Micro-Array NORmalization of Previous: 6 Graphical representations Contents
> dir.in <- system.file("data", package = "MANOR")
> spot.names <- c("LogRatio", "RefFore", "RefBack", "DapiFore",
+ "DapiBack", "SpotFlag", "ScaledLogRatio")
> clone.names <- c("PosOrder", "Chromosome")
> edge <- import(paste(dir.in, "/edge.txt", sep = ""), type = "spot",
+ spot.names = spot.names, clone.names = clone.names, add.lines = TRUE)
[1] "number of lines does not match array design: adding empty lines..."
> data(flags) > data(spatial) > local.spatial.flag$args <- alist(var = "ScaledLogRatio", by.var = NULL, + nk = 5, prop = 0.25, thr = 0.15, beta = 1, family = "gaussian") > flag.list <- list(spatial = local.spatial.flag, spot = spot.corr.flag, + ref.snr = ref.snr.flag, dapi.snr = dapi.snr.flag, rep = rep.flag, + unique = unique.flag) > edge.norm <- norm.arrayCGH(edge, flag.list = flag.list, FUN = median, + na.rm = TRUE) [1] "spatial" [1] "mean of unbiased zone : -0.0231566395663957" [1] "Spatial bias has been detected" zone.number mu effectif effectif.cumul frequency.cumul biased.zone 4 5 0.467833333 66 66 0.00918964 1 3 4 0.045546490 1581 1647 0.22932331 0 5 3 0.004946157 2693 4340 0.60428850 0 1 2 -0.034216274 1868 6208 0.86438318 0 2 1 -0.079646817 974 7182 1.00000000 0 [1] "spot" [1] "ref.snr" [1] "dapi.snr" [1] "rep" [1] "unique" > edge.norm <- sort.arrayCGH(edge.norm, position.var = "PosOrder")
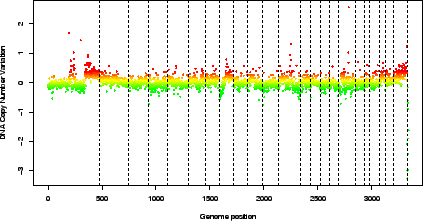
> report.plot(edge.norm, chrLim = "LimitChr", zlim = c(-1, 1)) 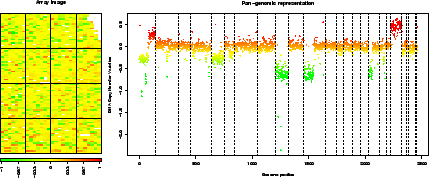 |
> profileCGH <- as.profileCGH(edge.norm$cloneValues)
> profileCGH <- daglad(profileCGH, smoothfunc = "lawsglad", lkern = "Exponential",
+ model = "Gaussian", qlambda = 0.999, bandwidth = 10, base = FALSE,
+ round = 2, lambdabreak = 6, lambdaclusterGen = 20, param = c(d = 6),
+ alpha = 0.001, msize = 5, method = "centroid", nmin = 1,
+ nmax = 8, amplicon = 1, deletion = -5, deltaN = 0.1, forceGL = c(-0.15,
+ 0.15), nbsigma = 3, MinBkpWeight = 0.35, verbose = FALSE)
[1] "Smoothing for each Chromosome"
[1] "Optimization of the Breakpoints"
[1] "Check Breakpoints Position"
> edge.norm$cloneValues <- as.data.frame(profileCGH)
> edge.norm$cloneValues$ZoneGNL <- as.factor(edge.norm$cloneValues$ZoneGNL)
> data(qscores)
> qscore.list <- list(smoothness = smoothness.qscore, var.replicate = var.replicate.qscore,
+ dynamics = dynamics.qscore)
> edge.norm$quality <- qscore.summary.arrayCGH(edge.norm, qscore.list)
> edge.norm$quality
name label score
1 LOCAL_SMOOTHNESS Local signal variability along the genome 0.021
2 VAR_REPLICATE Average variability among replicates 0.011
3 SIGNAL_DYNAMICS Dynamics of the DNA copy number variation 0.399
Function html.report generates an HTML file with key features of the normalization process: array image and genomic profile before and after normalization, spot-level flag report, and value of the quality criteria.
> html.report(edge.norm, dir.out = ".", array.name = "an array with local bias", + chrLim = "LimitChr", light = FALSE, pch = 20, zlim = c(-2, + 2), file.name = "edge")
The results of the previous command can be viewed in the file edge.html.
> spot.names <- c("Clone", "FLAG", "TEST_B_MEAN", "REF_B_MEAN",
+ "TEST_F_MEAN", "REF_F_MEAN", "ChromosomeArm")
> clone.names <- c("Clone", "Chromosome", "Position", "Validation")
> ac <- import(paste(dir.in, "/gradient.gpr", sep = ""), type = "gpr",
+ spot.names = spot.names, clone.names = clone.names, sep = "\t",
+ comment.char = "@", add.lines = TRUE)
[1] "number of lines does not match array design: adding empty lines..."
[1] "calculating array design..."
> ac$arrayValues$F1 <- log(ac$arrayValues[["TEST_F_MEAN"]], 2)
> ac$arrayValues$F2 <- log(ac$arrayValues[["REF_F_MEAN"]], 2)
> ac$arrayValues$B1 <- log(ac$arrayValues[["TEST_B_MEAN"]], 2)
> ac$arrayValues$B2 <- log(ac$arrayValues[["REF_B_MEAN"]], 2)
> Ratio <- (ac$arrayValues[["TEST_F_MEAN"]] - ac$arrayValues[["TEST_B_MEAN"]])/(ac$arrayValues[["REF_F_MEAN"]] -
+ ac$arrayValues[["REF_B_MEAN"]])
> Ratio[(Ratio <= 0) | (abs(Ratio) == Inf)] <- NA
> ac$arrayValues$LogRatio <- log(Ratio, 2)
> gradient <- ac
> data(spatial) > data(flags) > flag.list <- list(local.spatial = local.spatial.flag, spot = spot.flag, + SNR = SNR.flag, global.spatial = global.spatial.flag, val.mark = val.mark.flag, + position = position.flag, unique = unique.flag, amplicon = amplicon.flag, + chromosome = chromosome.flag, replicate = replicate.flag) > gradient.norm <- norm.arrayCGH(gradient, flag.list = flag.list, + FUN = median, na.rm = TRUE) [1] "local.spatial" [1] "mean of unbiased zone : 8.39530436452067" [1] "There is no spatial bias" zone.number mu effectif effectif.cumul frequency.cumul biased.zone 1 7 8.693635 510 510 0.05083732 0 2 6 8.606773 626 1136 0.11323764 0 3 5 8.494905 1083 2219 0.22119219 0 4 4 8.469920 2510 4729 0.47139155 0 5 3 8.409388 2520 7249 0.72258772 0 6 2 8.349206 2222 9471 0.94407895 0 7 1 8.180788 561 10032 1.00000000 0 [1] "spot" [1] "SNR" [1] "global.spatial" [1] "val.mark" [1] "position" [1] "unique" [1] "amplicon" [1] "chromosome" [1] "replicate" > gradient.norm <- sort.arrayCGH(gradient.norm)
> profileCGH <- as.profileCGH(gradient.norm$cloneValues)
> profileCGH <- daglad(profileCGH, smoothfunc = "lawsglad", lkern = "Exponential",
+ model = "Gaussian", qlambda = 0.999, bandwidth = 10, base = FALSE,
+ round = 2, lambdabreak = 6, lambdaclusterGen = 20, param = c(d = 6),
+ alpha = 0.001, msize = 5, method = "centroid", nmin = 1,
+ nmax = 8, amplicon = 1, deletion = -5, deltaN = 0.1, forceGL = c(-0.15,
+ 0.15), nbsigma = 3, MinBkpWeight = 0.35, verbose = FALSE)
[1] "Smoothing for each Chromosome"
[1] "Optimization of the Breakpoints"
[1] "Check Breakpoints Position"
> gradient.norm$cloneValues <- as.data.frame(profileCGH)
> gradient.norm$cloneValues$ZoneGNL <- as.factor(gradient.norm$cloneValues$ZoneGNL)
> data(qscores)
> qscore.list <- list(smoothness = smoothness.qscore, var.replicate = var.replicate.qscore,
+ dynamics = dynamics.qscore)
> gradient.norm$quality <- qscore.summary.arrayCGH(gradient.norm,
+ qscore.list)
> gradient.norm$quality
name label score
1 LOCAL_SMOOTHNESS Local signal variability along the genome 0.032
2 VAR_REPLICATE Average variability among replicates 0.050
3 SIGNAL_DYNAMICS Dynamics of the DNA copy number variation 0.294
Function html.report generates an HTML file with key features of the normalization process: array image and genomic profile before and after normalization, spot-level flag report, and value of the quality criteria.
> html.report(gradient.norm, dir.out = ".", array.name = "an array with spatial gradient", + chrLim = "LimitChr", light = FALSE, pch = 20, zlim = c(-2, + 2), file.name = "gradient")
The results of the previous command can be viewed in the file gradient.html.