MICSA: ChIP-Seq data Analysis by de novo motif identification
MICSA is package for the identification of transcription factor binding sites in ChIP-Seq data developed by the Computational Systems Biology of Cancer group at the Bioinformatics Laboratory of Institut Curie (Paris).
Cite: Boeva V, Surdez D, Guillon N, Tirode F, Fejes AP, Delattre O, Barillot E. De novo motif identification
improves the accuracy of predicting transcription factor binding sites in ChIP-Seq data analysis. Nucleic Acids Res. 2010 Jun 1;38(11):e126. Epub 2010 Apr 7.
Download the article: HTML, PDF.
MICSA HOWTOs:
1. Installing the MICSA package
2. Running MICSA on NRSF test data
3. Running MICSA on EWS-FLI1 dataset
4. Reading MICSA's output
Contact:
Valentina Boeva: micsa@curie.fr
phone: +33.1.56.24.69.31
Distributions:
MICSA is distributed as an open source application. However it calls MEME which is free only for non-commercial use. Please check MEME license before using
- current release of MICSA without MEME and FindPeaks [ download ]
- precompiled versions for Linux64, Linux32 and Windows OS
Test data:
- NRSF human ChIP-Seq data (Neural-restrictive silencing factor, hg18) and control data (RX_noIP, hg18) [ download ] (alignment by Eland)
- EWS/FLI1 human ChIP-Seq data (Oncoprotein of Ewing sarcoma, hg18) and control data (MON, hg18) (alignment by MAQ)
Installation:
- Download and install the latest version of MEME from the MEME website. MEME is free only for non-commercial use. Please check the MEME license before using.
- Add the directory with meme.bin ("meme_your_version\src"?) to your PATH.
- type export PATH=$PATH:/YOUR PATH TO meme.bin
In LINUX make meme.bin runnable by chmod 755 meme.bin
- Download the MICSA program: MICSA. The archive includes a version of FindPeaks.jar
- If you want to run the latest version of FindPeaks, download and install the latest version of FindPeaks from the FindPeaks website
Running MICSA on NRSF test data
- Download ChIP and control data for NRSF:
- Unpack ChIP and control data:
type unzip *.zip
instead of '*.zip' put the name of your zip file
- Download and unpack genome sequences in fasta format. hg18 (for NRSF test), mm9 (in case if you have mouse data)
- Launch the GUI for MICSA pipeline
type java -Xmx1500m -jar micsaGUI.jar
or run MICSA pipeline in command line.
MICSA by graphical interface
Launch the GUI and fill in the form as shown in the example:
Click "Check input information"
Click "Start"
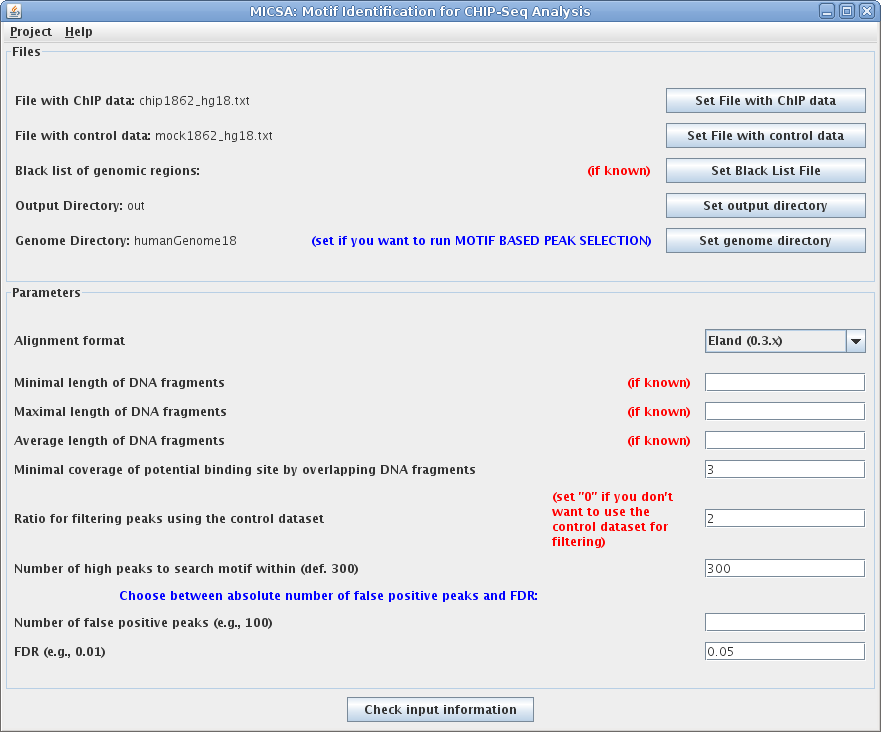
Files and parameters in this example:
- File with ChIP data: chip1862_hg18.txt file with ChIP-Seq data for NRSF, aligned by Eland to the Human Genome assembly 18 (Mar.2006).
- File with control data: mock1862_hg18.txt file with control data, aligned by Eland to the Human Genome assembly 18 (Mar.2006).
Both files can be downloaded here.
- Black list of genomic regions: hg18_masked_Centr (Download hg18_masked_Centr) file with positions of pericentromeric repeats (e.g., Alpha-satellites) in hg18 downloaded from UCSC Genome Browser.
You can use other files for masking. For example: hg18 satellites
, hg18 whole pericentromeric and heterochomatin regions.
- Output Directory select your output directory. If the specified directory does not exist, it will be created.
- Genome Directory: hg18 directory with human genome files. The human genome (assembly Mar.2006) can be downloaded here.
- Alignment format - Eland (0.3.x) here you need to specify the alignment format for your data. The NRSF data are in format Eland.
- Minimal, maximal and average length of DNA fragment optional parameters. Set them if your know the length distribution of the DNA fragments you sequenced.
- Minimal coverage of potential binding site by overlapping DNA fragments set how many overlapping DNA fragments you need to keep a peak. This value is roughly equal to the minimal number of mapped DNA reads in a window (the minimal value is 3).
- Ratio for filtering peaks using the control data in the case when a peak in the ChIP data overlaps with a peak in the
control data, the former will be discarded if the height of peak from ChIP data devided by the hight of peak in the control data is smaller than the specified ratio.
- Number of false positive peaks Number of false peaks you allow in the output. With 120 false positives you can find about 5000 peaks. Alternatively, you can specify a false discovery rate, which is the ratio between the evaluated number of false positive binding sites and the total number of predicted binding sites.
MICSA by command line
- Run FindPeaks on your data ChIP and control data
- Create wig directory in /YOUR_OUTPUT_DIRECTORY/ :
type in CygWin or in your Linux shell:
mkdir /YOUR_OUTPUT_DIRECTORY/wig
- type in CygWin or in your Linux shell (don't forget to change paths to your directories):
java -Xmx2G -jar PATH_TO_FINDPEAKS/FindPeaks.jar -aligner eland -duplicatefilter -input PATH/out_chip/*.part.eland.gz -name chip -output YOUR_OUTPUT_DIRECTORY/wig -dist_type 1 -minimum 3
java -Xmx2G -jar PATH_TO_FINDPEAKS/FindPeaks.jar -aligner eland -duplicatefilter -input PATH/out_control/*.part.eland.gz -name control -output YOUR_OUTPUT_DIRECTORY/wig -dist_type 1 -minimum 1
Read the FindPeaks Manual for more options.
FindPeaks can output either the entire data track or provide the data track split by chromosome. If you have a large dataset, it is best to upload only chromosomes that you are interested in.
UCSC genome browser does not accept large files,
so attempting to load an excessively large wig file may cause the upload to fail altogether.
It is suggested that you use the by-chromosome option, triggered by the -one_per
flag on the command line for FindPeaks.
UCSC Genome Browser sessions will expire after about 3 days of inactivity, however, you may keep a session alive longer by "refreshing" the current session (by hitting "referesh" button or by zooming in or out).
- Filter out peri-centromeric or satellite regions.
The following masking data sets are available for use in MICSA:
- Create summary about peak distribution in ChIP and control data
java Summary -f chip_triangle_standard.peaks -c control_triangle_standard.peaks -r 0.735371287
| Mandatory parameters:
|
| -f | input file
|
| -c | control file
|
| Optional parameters:
|
| -r | (default 1) ratio between number of tags in ChIP and that in control data
|
Information about tag number can be fount in "meta_info.txt" file of FindPeaks
- Filter out peaks occurring both in ChIP and Control data:
java FilterPeaks -f chip_triangle_standard.peaks -c control_triangle_standard.peaks -t 3.5
| Mandatory parameters:
|
| -f | input file
|
| -c | control file
|
| Optional parameters:
|
| -w | (default 0) flanks to add to window length in contol data
|
| -v | (default 2) enrichment fold over control data
|
- Run MICSA.jar
java -jar -Xmx2G PATH_TO_MICSA/micsa.jar -name micsaTest -f wig/chip_triangle_standard.peaks
-n 50 -o "outputDir" -l "wig/FindPeaksSummary.txt" -g "PATH/humanGenome" -w "wig/chip_triangle_standard.wig.gz"
| Mandatory parameters:
|
| -f | input file
|
| -n | maximal number of expected false positives
|
| -fdr | maximal value of FDR (then "-n" option should be omitted)
|
| -o | path to the output directory
|
| -l | file with summary
|
| -g | path to the genome directory
|
| -w | file with wig file produced by FindPeaks
|
Running MICSA on EWS-FLI1 dataset
By graphical interface
- Download ChIP and
control data for EWS-FLI1.
- Download and unpack genome sequences in fasta format. hg18 (for EWS-FLI1), mm9 (in case if you have mouse data)
- Run GUI for MICSA pipeline
type java -Xmx1500m -jar micsaGUI.jar
or run MICSA pipeline in command line.
Fill the form as in the example:
Click "Check input information"
Click "Start"
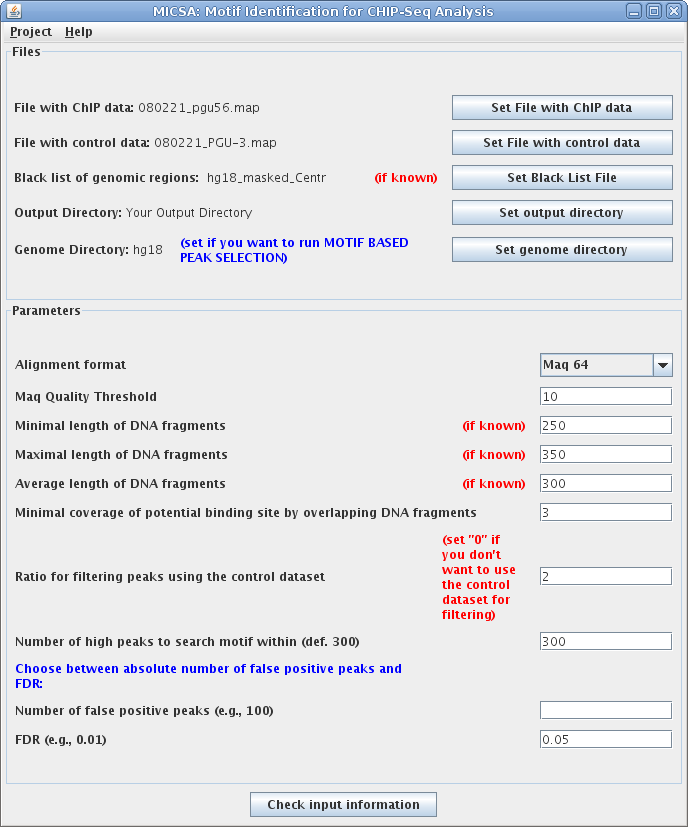
Files and parameters in this example:
- File with ChIP data: 080221_pgu56.map File with ChIP-Seq data for EWS-FLI1 (download), aligned by Maq to the Human Genome assembly 18 (Mar.2006).
- File with control data: 080221_PGU-3.txt File with control data ( download), aligned by Maq to the Human Genome assembly 18 (Mar.2006).
- Black list of genomic regions: hg18_masked_Centr (Download hg18_masked_Centr) file with positions of pericentromeric repeats (e.g., Alpha-satellites) in hg18 downloaded from UCSC Genome Browser.
You can use other files for masking. For example: hg18 satellites
, hg18 whole pericentromeric and heterochomatin regions.
- Output Directory Select your output directory. If the specified directory does not exist, it will be created.
- Genome Directory: hg18 Directory with human genome files. The human genome (assembly Mar.2006) can be downloaded here.
- Alignment format - Maq 64 Here you need to specify the alignment format for your data. The EWS-FLI1 data are in format Maq 64.
- Minimal, maximal and average length of DNA fragment Optional parameters. Set them if your know the length distribution of the DNA fragments you sequenced.
- Minimal coverage of potential binding site by overlapping DNA fragments Set how many overlapping DNA fragments you need to keep a peak. This value is roughly equal to the minimal number of mapped DNA reads in a window (the minimal value is 3).
- Ratio for filtering peaks using the control data In the case when a peak in the ChIP data overlaps with a peak in the
control data, the former will be discarded if the height of peak from ChIP data devided by the hight of peak in the control data is smaller than the specified ratio.
- Number of peaks to search motif within Maximal number of peak which will be used by MEME to call motifs
- Number of false positive peaks Leave blank if you want to use FDR instead.
- FDR False discovery rate you allow in the output.
MICSA's output
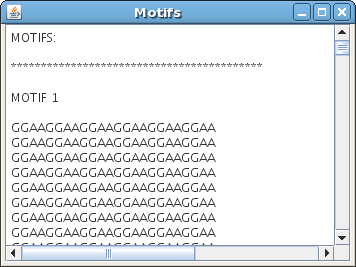
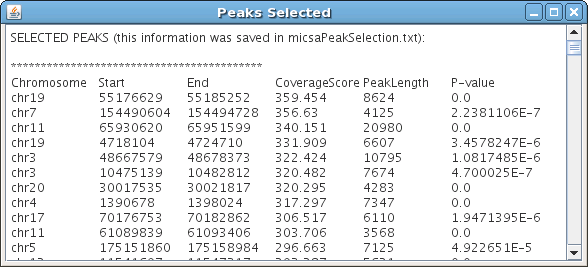
MICSA outputs information about motifs ("motifs.txt" file) and a file with peak coordinates.
Information for each peak:
- chromosome
- start
- end
- maximal depth of coverage by overlapping DNA fragments
- length
- score
Score is small for high confidence peaks.
In GUI version you will see your results appear in a window like this:
Last modified: June 24 2009