Tatiana Popova, Elodie Manie, Dominique Stoppa-Lyonnet, Guillem Rigaill, Emmanuel Barillot and Marc-Henri Stern
Genome Biology 2009, 10:R128
Introduction
Single nucleotide polymorphism (SNP) arrays are presently one of the most efficient technologies for the identification of genetic alterations. SNP-arrays simultaneously define copy number changes and allelic imbalances (including LOH) occurring in a tumor, at high resolution and throughout the whole genome (examples of Illumina and Affymetrix SNP array profile).
Problems in analysis of complex cancer genomes
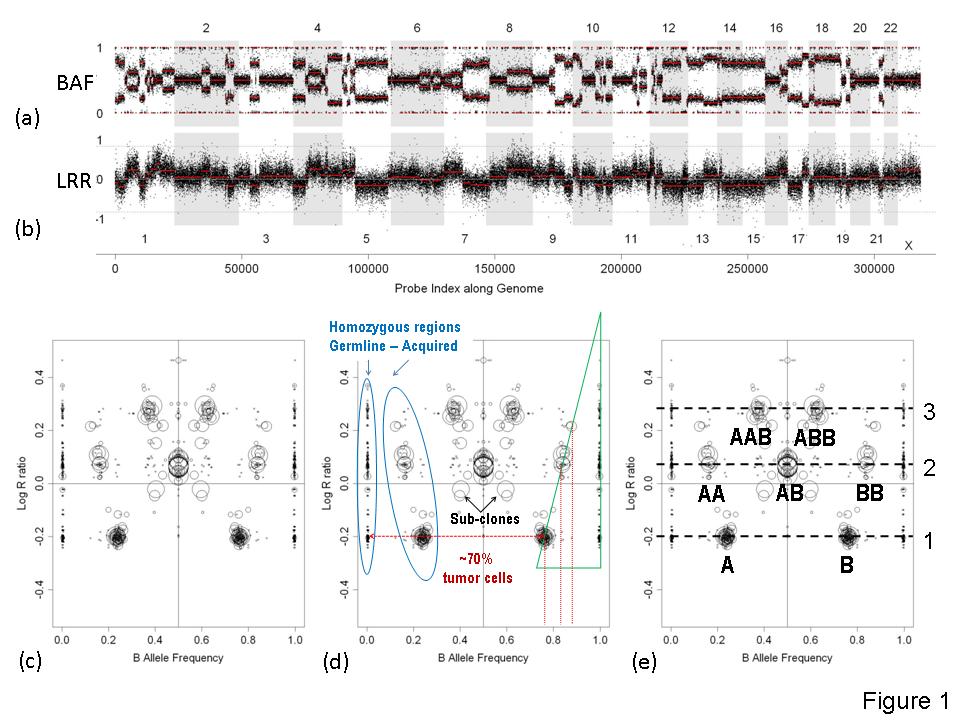
- Cancer sample is often contaminated by normal stromal cells, which add normal DNA to measured material. Normal DNA addition smooth changes in Log R ratio and compress B allele frequencies towards 0.5
- If tumor sample contains sub-clones measured profile would have intermediate levels, which are hard to attribute
- Experimental noise makes Log R ratio profile much less variant than it is implied by theory: threshold approach does not work
- Tumor genome could be near-tetraploid. Attribution of gain/loss status is then dependent on ploidy. We need underlying ploidy recognition, etc
We describe a method for automatic detection of absolute segmental copy numbers and genotype status in complex cancer genome profiles measured by SNP arrays. The method is based on pattern recognition of segmented and smoothed copy number and allelic imbalance profiles.
This method is based on SNP-array data formalization that we have called Genome Alteration Print (GAP). GAP of a tumor sample summarizes segmented CNV and allelic imbalance profiles into a list of segments, characterized by two corresponding averages. GAP visualization reveals the overall genomic ploidy of tumors, pinpoints the possible normal status (reference point for gain and loss), shows the level of contamination, indicates subclones, and generally characterizes the tumor genome. The model GAP built on theoretical distribution of CNV and allelic imbalances provides interpretation for a tumor GAP and serves as a basis for automatic recognition of copy number and genotype of each segment. The method performs well even for poor quality data, low tumor content, and highly rearranged tumor genomes.
Automatic recognition of segmental copy numbers and genotypes
GAP pattern can be easily mined by automatic procedures. This procedure includes
- recognition of a GAP pattern and
- assignment of segmental copy numbers and genotypes to corresponding tumor genome based on this pattern.
Illustration
High-resolution figures and supplementary data from the article:
References
- Popova T, Manie E, Stoppa-Lyonnet D, Rigaill G, Barillot E, Stern MH. Genome Alteration Print (GAP): a tool to visualize and mine complex cancer genomic profiles obtained by SNP arrays. Genome Biol. 2009;10(11):R128.
- Popova T, Boeva V, Manie E, Rozenholc Y, Barillot E, Stern, MH. Analysis of Somatic Alterations in Cancer Genome: From SNP Arrays to Next Generation Sequencing. Genomics I – Humans, Animals and Plants. ISBN: 978-1-477554-91-3. iConcept Press.
- Popova T, Manie E, Stern M.-H. Genomic Signature of Homologous Recombination Deficiency in Breast and Ovarian Cancers. Bio-Protocol.
Downloads
Contact information