

|
|
Coordinator: Patrick Poullet.
Developers: Patrick Poullet and Sabrina Carpentier.
This project is developed in collaboration with the Curie
Mass Spectrometry Group headed by Damarys Loew.
Now that a significant number of genomes (including the human genome) have been sequenced and that the expression level of each gene can been monitored using various DNA chip technologies, researchers are turning to analysing the distribution profile of the thousands of proteins encoded by these genomes (proteomes). Mass Spectrometry (MS) has increasingly become the method of choice to achieve this challenging task.
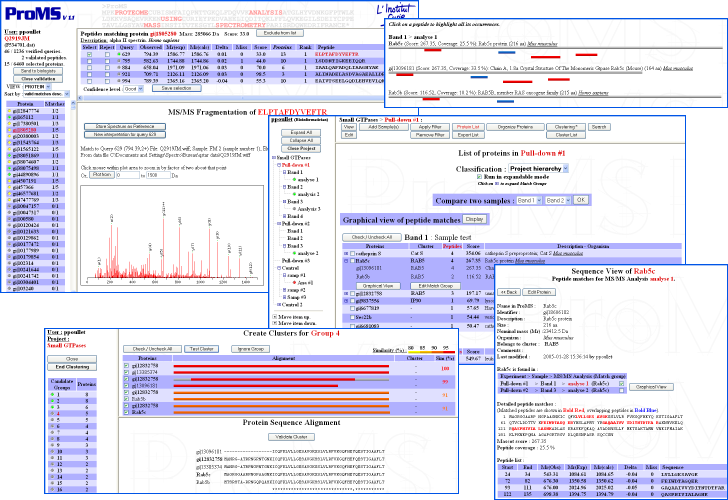
MS-based proteomics combines peptide ionisation and fragmentation technologies with the availability of gene sequence databases to identify the protein content of complex biological samples such as entire cells. During a typical MS analysis performed at the Curie Institute, hundreds of peptide fragmentation spectra are generated and interpreted using the specialised software MASCOTTM (Matrix Sciences). A single spectrum can be interpreted into up to 10 compatible peptide sequences which in turn correspond to several proteins. Manual validation of the accurate interpretations by MS specialists (massists) is therefore necessary in order to correctly identify the protein content of a biological sample. Although necessary, these currations become a bottle-neck when a large number of samples must be analysed. In addition, MASCOTTM is designed neither to coherently manipulate results from multiple MS analyses nor to allow efficient data sharing between different laboratories.
In order to solve these issues, we have developed ProMS (Proteome Analysis using Mass Spectrometry). The ProMS web server is ideally suited for the low/medium throughput MS requirement of the Curie Institute.
ProMS functions can be divided into two main features:
- It allows the user to centralise and organise data from several MS analyses in a single database.
Massists and biologists can access the database through a web interface to upload, visualise, edit and retrieve their data.
- ProMS optimises manual curation of the spectrum interpretations by improving data visualisation
and comparison during the validation process. Consequently, only the validated peptide sequences
and corresponding proteins become accessible to the biologists who provided the protein samples analysed.
ProMS was developed in Perl, HTML and Javascript and uses Oracle or MySQL database systems.
ProMS is freely available for academic users assuming they already possess a MASCOTTM licence (contact: proms@curie.fr).